Sorry for schedule change.
Date: 12/ 7 (Friday), 2018, 13:30 -16:50 Venue: 人文館遠距會議室 |
|||
|
|||
|
13:30-13:50: 林仲彥 博士 (Chung-Yen Lin Ph.D.) 系統網路生物實驗室 (Lab of Systems Biology and Network Biology, http://eln.iis.sinica.edu.tw) 中央研究院資訊科學研究所 (Institute of Information Science, Academia Sinica) 內容:將簡介實驗室相關生物醫學巨量資料研究、以及公開發表與次世代定序相關之模式生物,與非模式生物多維體分析平台生物資訊系統平台、DOCKER程式與相關人工智慧運用等。研究團隊希望,透過本次的工作坊,鎖定轉錄體與全基因體甲基化的分析,從原始定序產出到後端生物意義解析,讓使用者快速瞭解與運用我們所開發的資訊工具,來處理巨量生物序列,並減低大數據與資訊技術門檻所帶來的障礙,擷取其中的關鍵差異資料,並進行與個人專業領域相關的深度分析。 This brief will show our recently research works about biomedical big data and their applications. Meanwhile, we will show you those published web applications, databases, DOCKER images and frameworks in AI/Deep Learning available for public. **本課程部分工具程式將使用DOCKER平台,如自行攜帶筆電,請先安裝相關套件,安裝資訊請參閱連結 (WINDOWS 10部分,僅支援PRO及學術版,家用版無法使用) (Mac),本次課程也會示範安裝過程。
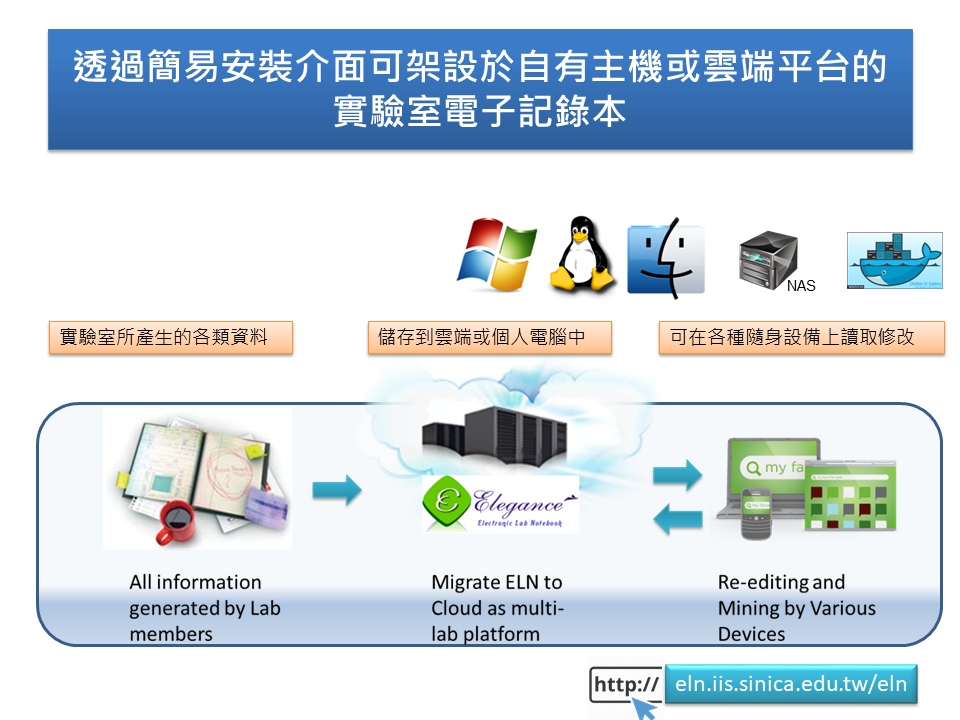
13:50 - 14:50 黃智偉 先生 (Mr. Chi-Wei Huang) 中央研究院資訊科學研究所 (Institute of Information Science, Academia Sinica) 內容: 電子實驗室記錄本之運用與維護 將實驗室中所產生的各種紀錄與大量數據,以電子化方式儲存於網路伺服器/一般桌上型/筆記型電腦/雲端/NAS,可藉由一般電腦、筆記型電腦與移動式裝置如智慧型手機與平板電腦等,以一般的網路瀏覽器就可以來存取實驗相關內容與研究數據,以通用網頁編輯的高親和性介面,協助使用者管理、分享、搜尋、備份、列印,及與國內外研究伙伴線上討論相關問題,讓實驗室的眾多智慧、想法與研究歷程得以紀錄回顧與交互串連,不受時空與人員異動的影響,讓實驗室的智慧結晶得以快速存取與整合,並隨時上傳新的想法與心得,即時分享給研究伙伴,加速研究的進展。 本系統以開放源碼(Open Source)為基礎,並融入web 2.0的精神,整合網路工具(durpal, apache, Ajax)、資料庫(mySQL)與程式語言(php, Java, C++),可以在不同作業系統 (Linux, Windows, Mac)或是雲端平台上,透過高親和性的簡易介面進行安裝,無須複雜的資訊技術協助,便能同時快速建置屬於自己或是中小型研究團隊所需的資料交換分享平台及對外網站。目前也已製作DOCKER影像檔,除了可以直接於Linux或是一般雲端平台設定外,使用者也可以先行安裝DOCKER 平台於WINDOWS/MAC/ NAS,無須繁複的程式安裝,就能快速建置可供多人使用的實驗室資訊分享平台。 除了可提供各類型實驗資料(文字、數據、檔案、圖像及影音等)的儲存列表、增刪修訂、搜尋分類、專案與參與人員管理、瀏覽列印、安全防護以及自動複製備份等功能,並能進一步協助建置資料交換分享、線上討論、即時編修、實驗報告版本修改紀錄管理、實驗室耗材藥品資源管理與數位簽章等機制。目前此一系統內建中文、日文與英文之使用介面,並提供包括雲端平台等多種版本,透過對Web介面的優化,除了一般的電腦外,也讓不同的移動設備如智慧型手機、平板電腦等,都能透過直覺的圖形化介面,在無時間與距離的限制下,存取與分享儲存在雲端的實驗室智慧,進而激盪出更多的研究火花與新的探討方向。 The hand-writing, paper-based recording way is not competent to keep data in increasing volumes and complexity, and is hard to make data sharing in a cooperating project among various disciplines and research communities. With more and more outputs generated with digital deluge from high throughput biology, a web platform we developed for knowledge repository with the functions like search, backup, reconstruction will be an important issue in current laboratories for daily records. Currently, we have developed the framework of pure web-based ELN which can be deployed on local PCs, local servers, NAS or clouds instead of high manpower required ELN server /client architecture. Meanwhile, users can access the ELN by any kinds of web browsers on various machine including mobile devices without the limitation of time and space. Thus, it can be shaped for managing thoughts and all kind of lab working logs/ experiment data for a single researcher, for a small research team to construct their own internet web service for public and intranet framework to manage experimental results, as well as a sharing working platform among labs.
平台: Electronic Laboratory Notebook (ELN) (影音簡介) |
|||
|
|||
|
15:00 - 15:40 謝秉恆 資訊工程師 (Ping-Heng Hsieh) 中央研究院資訊科學研究所 (Institute of Information Science, Academia Sinica) 內容:
由於定序技術的精進,生物序列多維體(基因體、轉錄體及甲基化等)巨量資料大量產生,如何對這些大數據進行快速計算及註解比對,並與原有資料相結合比較,找出生醫大數據背後所帶來的意義,已成為生物醫學研究中越來越重要的一個關鍵。然而定序所產生的大量原始數據,所帶來的數據處理、比對分析與備份儲存等課題,已是當代生物醫學的研究社群日常所面臨的一大問題。為了解決巨量生醫序列資料的解析,研究團隊在微軟公司贊助與科技部的支持下,於2014年開始建構轉錄體之線上資料分析雲端平台,並於今年完成原始資料到基因表現繁瑣流程的DOCKER虛擬平台雲端版本的建置,可協助研究人員,透過高親和圖像介面,以自有的想法,進行深入的分析,並對分析流程進行紀錄,以圖像化的方式來呈現基因表現差異、其所相關的調控功能、參與的代謝網路,及蛋白質互動網路的結果,來解析巨量序列片段背後的生物調控意義。
這樣的平台架構,除了應用於人類與小鼠的疾病研究與生理機制探討外,也可應用於非模式物種如日本鰻、龍膽石斑與白蟻等基因體的研究上,瞭解基因體、轉錄體、蛋白質體與甲基化之間的複雜調控關係。此系統轉錄體分析平台部分雛形,已於2016年透過中研院智財處的協助,以非專屬授權方式,技轉國內生物序列定序公司。
For alleviating the burden on processing massive NGS data, we create the workflows based on galaxy/ Docker to estimate expression profiling in RNA-seq. There are two components to complete the whole analysis. For the first part, we provide three kinds of ways to estimate the expression profiling shown as below via DOCKER.
After the data pre-processing done, the expression profiling can be submitted to MOLAS. MOLAS, Multi-Omics onLine Analysis System for model and non-model organisms, is a robust web application that can take gene expression data (FPKM/TPM) from different libraries as inputs, map these expressed genes with build-in annotations for further analyses and reveal biological meaning of the complex data in the intuitive interface. By integration of workflows on galaxy/Docker and MOLAS, researchers just need to find the computational resource to generate the profiling from the big raw reads they got. Then they can start to explore the biological meaning hidden inside the data via MOLAS without facing the IT hurdles and statistical challenges.
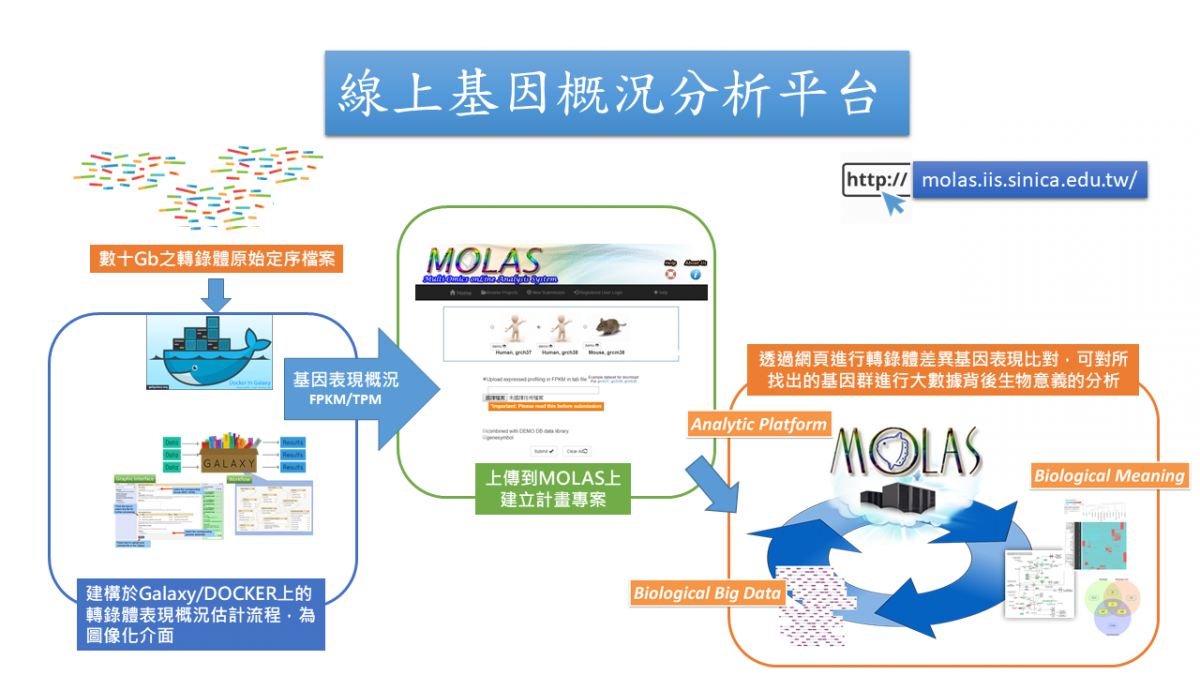 本分析流程可分析人類及小鼠之基因概況資料,然而因大數據處理運算需於雲端或高效能主機進行,而後續資料分析比對則於開發團隊的一般伺服器主機上,所以將整個流程分成兩大部分:
檔案下載: Test data [genome][gff][Rna_A.R1][Rna_A.R2][Rna_B.R1][Rna_B.R2]
15:50 - 16:50 Shu-Hwa Chen Ph.D., 陳淑華 博士 中央研究院資訊科學研究所 (Institute of Information Science, Academia Sinica) 內容: 本課程將簡略說明NGS技術和常見的應用,內容也將包括平台選擇,實驗設計的總體考量。 將簡介與展示全基因體甲基化線上分析平台(epi-MOLAS, 人類、小鼠與阿拉伯芥等模式生物),將以阿拉伯芥為範例 (TEA),引導使用者如何設計實驗,進行定序及資料的初步分析處理,透過線上系統的協助,以簡潔的介面來完成複雜的全基因體甲基化分析,並說明如何將甲基化分析結果與轉錄體資料結合,進行綜合分析。 In this session, we will brief the experimental design, raw data process and functional analyses for high throughput biology in next generation sequencing. DNA methylation is a crucial epigenomic mechanism in the biological system. Using whole genome bisulfite sequencing (WGBS) technology, the methylation status of cytosine sites can be revealed. However, performing WGBS data analysis is often complicated and challenging. To alleviate such difficulties, we integrated the WGBS data processing and downstream analysis into a two-phase approach, namely, EpiMOLAS. First, we packed a Docker container DocMethyl to deal with raw data processing, mapping, and methylation calling/ scoring to give the summary, mtable, of the whole genome methylation status by the gene. Next, mtables are uploaded to the web server EpiMOLAS_web for linking with gene annotation databases that enable rapid data retrieval and analyses. This two-phase combination of DocMethyl and EpiMOLAS_web solve methylome data analysis from raw reads processing to downstream analysis.
本分析流程可分析人類、小鼠及阿拉伯芥之全基因體甲基化概況資料,然而因大數據處理運算需於雲端或高效能主機進行,而後續資料分析比對則於開發團隊的一般伺服器主機上,所以將整個流程分成兩大部分:
|
|||


|
報名網頁: |